Roadmap for the development of medicinal products

To make new medicinal products available in clinical practice they need to be tested in animal studies and in human clinical trials, registered as medicinal products, and reimbursed by health insurance companies. This is a heavily regulated process. Laws and regulations for medicinal products need to be carefully considered throughout the whole development trajectory.
This roadmap shows the trajectory of drug development; from acquiring knowledge and target discovery to clinical studies and implementation in daily practice. It serves as a tool to give you an overview of the most important steps during the drug development process. Each step shows the goal of this step, the activities to be performed, and the end point of this step, which is the starting point of the next step. Also advice is given which regulatory body should be contacted.
(click here to download full roadmap - PDF)
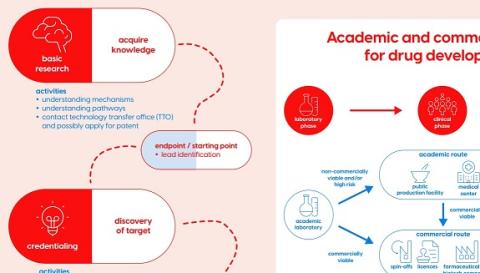
Medicinal products include different types of medicines; small molecules (i.e. traditional pharmaceuticals), biological medicinal products (e.g. monoclonal antibodies), and cell and gene therapies (i.e. Advanced Therapy Medicinal Products). Like other innovations these need to be investigated first in two different animal models (i.e. preclinical research), and after sufficient evidence of safety, efficacy, and mechanism of action has been gathered, in human clinical trials.
For first-in-human studies and all subsequent trials, approval of the clinical trial is required based on the Investigational Medicinal Product Dossier (IMPD). In the Netherlands, IMPD’s are reviewed by medical ethics committees and/or the Dutch Central Committee on Research Involving Human Subjects (Centrale Commissie Mensgebonden Onderzoek, CCMO). As of January 21, 2022, the European Clinical Trial Regulation was implemented, with a transition period of three years. It aims to harmonise clinical trial evaluation, conduct and oversight in the European Union (EU). This means the evaluation process will change for all trials. More information on the changes, new process, and training can be found on the CCMO website (see links below).
All new oncology medicinal products are evaluated through the centralised authorisation procedure of the European Medicines Agency (EMA) for marketing authorization. If the outcome is positive the new medicinal product can be marketed in all EU Member States. The EMA bases its decision-making for marketing authorization on a risk/benefit analysis of the entire dossier that has been established throughout all phases of development. The rules and requirements for marketing authorization are very strict.
It is strongly recommended to seek advice from regulatory bodies as early as the preclinical phase of development, in order to meet demands of authorisation later on. Informal guidance, prior to formal scientific advice meetings, is provided by the Dutch Medicines Evaluation Board (College ter Beoordeling van Geneesmiddelen, CBG) or the Innovation task force of the EMA. Formal guidance can be provided through scientific advice meetings with CBG or other national regulatory agencies and/or the EMA. They assess the regulatory feasibility of study designs and methodologies and provide guidance on regulatory pathways or schemes that would be suitable for the product, among others. For instance, the EMA has specific designations for orphan drugs (ODD) and priority medicines (PRIME). Such designations provide benefits for developers, such as scientific advice without fees. EMA also provides a qualification procedure of new methodologies that support drug development, such as novel biomarkers.
For reimbursement by health insurance companies, Health Technology Assessments (HTA) are conducted by the Care Institute Netherlands (Zorginstituut Nederland, ZINL), based on a cost-effectiveness analysis as supplied by the manufacturer. In other EU countries, the national HTA bodies perform their own cost-effectiveness analysis that is tied to their national health care system. It is critical for new medicinal products to fit within the existing health care system and available treatments to be reimbursed. Therefore, it is strongly recommended to perform early HTA analyses as early as the preclinical phase of development, and to seek advice from the Care Institute Netherlands (ZINL) to discuss requirements and routes to clinical practice.
Relevant regulatory links are provided here:
- CBG – Informal and formal scientific advice: https://www.cbg-meb.nl/onderwerpen/hv-wetenschappelijk-en-regulatoir-advies. Contact: [email protected]
- CCMO - of IMPD: https://www.ccmo.nl/onderzoekers/geneesmiddelenonderzoek-ctr/voorbereiding-ctr/onderzoeksdossier-deel-i/investigational-medicinal-product-dossier-impd
- CCMO – Clinical drug development: https://www.ccmo.nl/onderzoekers/wet-en-regelgeving-voor-medisch-wetenschappelijk-onderzoek/wetten/europese-verordening-voor-klinisch-geneesmiddelenonderzoek-ctr
- DCRF – Clinical oncology research: https://dcrfonline.nl/handige-links-en-documenten/
- EMA - Innovation task force: https://www.ema.europa.eu/human-regulatory/research-development/innovation-medicines
- EMA - Start page for Academia: https://www.ema.europa.eu/partners-networks/academia. Contact: [email protected] or contact form at https://www.ema.europa.eu/
- EMA - Scientific advice: https://www.ema.europa.eu/human-regulatory/research-development/scientific-advice-protocol-assistance
- EMA - Orphan Drug Designation: https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
- EMA - PRIME: https://www.ema.europa.eu/human-regulatory/research-development/prime-priority-medicines
- EMA - Novel methodology advice & qualification: https://www.ema.europa.eu/human-regulatory/research-development/scientific-advice-protocol-assistance/qualification-novel-methodologies-medicine-development-0
- ZINL – Beoordeling van geneesmiddelen: https://www.zorginstituutnederland.nl/over-ons/werkwijzen-en-procedures/adviseren-over-en-verduidelijken-van-het-basispakket-aan-zorg/beoordeling-van-geneesmiddelen


